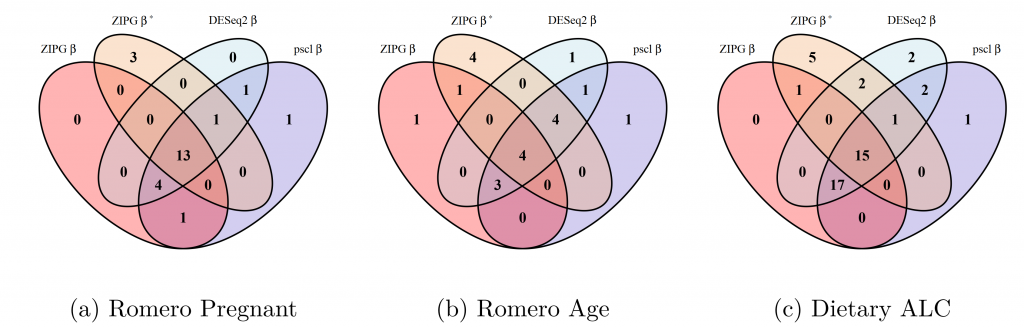
2023年3月25日,由清华大学统计学研究中心、清华大学交叉信息研究院与微软亚洲研究院联合主办的机器学习基础理论研讨会(Workshop on Machine Learning Theory and Foundations)在微软大厦顺利举办。本次研讨会邀请15名全球范围内机器学习领域的优秀学者,围绕最新的研究成果进行主题报告与分享,并针对当前机器学习领域的前沿问题展开深入交流与探讨。
2023年3月25日,由清华大学统计学研究中心、清华大学交叉信息研究院与微软亚洲研究院联合主办的机器学习基础理论研讨会(Workshop on Machine Learning Theory and Foundations)在微软大厦顺利举办。本次研讨会邀请15名全球范围内机器学习领域的优秀学者,围绕最新的研究成果进行主题报告与分享,并针对当前机器学习领域的前沿问题展开深入交流与探讨。  微软亚洲研究院的张辉帅研究员代表主办单位致辞。他指出,机器学习近年来的发展日新月异,产生了大量应用成果,而在领域前沿的研究中,机器学习理论发挥着至关重要的作用。同时,张博士介绍了本次会议的组织者,来自清华大学统计学研究中心的杨朋昆助理教授、清华大学交叉信息研究院张景昭助理教授以及微软亚洲研究院的孙丽君、杨格、张辉帅研究员。
微软亚洲研究院的张辉帅研究员代表主办单位致辞。他指出,机器学习近年来的发展日新月异,产生了大量应用成果,而在领域前沿的研究中,机器学习理论发挥着至关重要的作用。同时,张博士介绍了本次会议的组织者,来自清华大学统计学研究中心的杨朋昆助理教授、清华大学交叉信息研究院张景昭助理教授以及微软亚洲研究院的孙丽君、杨格、张辉帅研究员。  来自MosaicML研究团队的Jonathan Frankle研究员首先进行了报告。他的分享聚焦于神经网络的快速训练算法。当前,以GPT系列为代表的超大规模神经网络在实践中表现出了极好的性能,但在实践层面,超大规模神经网络的训练耗时过长且成本十分昂贵,没有巨额预算的研究人员和企业很难跟上前沿的发展。Frankle介绍了其团队在MosaicML团队对于神经网络训练算法的最近研究成果,包括通用理论、细节的改进与实践经验等。改进后的算法将标准计算机视觉模型的训练时间减少了5-7倍,标准语言模型的训练时间减少了2-3倍。
来自MosaicML研究团队的Jonathan Frankle研究员首先进行了报告。他的分享聚焦于神经网络的快速训练算法。当前,以GPT系列为代表的超大规模神经网络在实践中表现出了极好的性能,但在实践层面,超大规模神经网络的训练耗时过长且成本十分昂贵,没有巨额预算的研究人员和企业很难跟上前沿的发展。Frankle介绍了其团队在MosaicML团队对于神经网络训练算法的最近研究成果,包括通用理论、细节的改进与实践经验等。改进后的算法将标准计算机视觉模型的训练时间减少了5-7倍,标准语言模型的训练时间减少了2-3倍。  普林斯顿大学的Boris Hanin助理教授报告了深度神经网络贝叶斯插值的最新研究成果。Hanin的结果表明,在贝叶斯估计的视角下,无限深度的线性神经网络可以基于普遍、未知的先验进行最优后验的计算。在技术细节方面,他提出了一个新的缩放参数,可以对后验分布的复杂性和有效深度进行较好的控制。
普林斯顿大学的Boris Hanin助理教授报告了深度神经网络贝叶斯插值的最新研究成果。Hanin的结果表明,在贝叶斯估计的视角下,无限深度的线性神经网络可以基于普遍、未知的先验进行最优后验的计算。在技术细节方面,他提出了一个新的缩放参数,可以对后验分布的复杂性和有效深度进行较好的控制。  伊利诺伊大学香槟分校的Maxim Raginsky教授的报告主题为镜像朗之万动力学(Mirror Langevin Dynamics)中的变分法则。镜像下降法是一种经典的对偶凸优化方法,它通过定义一个距离生成势函数(distance-generating potential function)来刻画优化问题的几何结构。Raginsky教授的最新工作给出了镜像下降及其随机版本的变分公式。隐式正则化的表述可以对该结果进行较好的解释。
伊利诺伊大学香槟分校的Maxim Raginsky教授的报告主题为镜像朗之万动力学(Mirror Langevin Dynamics)中的变分法则。镜像下降法是一种经典的对偶凸优化方法,它通过定义一个距离生成势函数(distance-generating potential function)来刻画优化问题的几何结构。Raginsky教授的最新工作给出了镜像下降及其随机版本的变分公式。隐式正则化的表述可以对该结果进行较好的解释。  斯坦福大学的李志远博士后分享了对于锐度感知最小化(Sharpness-Aware Minimization, SAM) 的研究成果。SAM是一种重要的神经网络正则化方法,通过在神经网络优化中添加对锐度概念的惩罚项来实现更好的泛化性能。最新的结果通过对锐度概念的明确,在理论层面揭示了SAM的内在机制。特别地,SAM中的两个近似步骤分别都会导致不准确的中间结果,但在全梯度方法下,它们的组合意外地表现了良好的效果。
斯坦福大学的李志远博士后分享了对于锐度感知最小化(Sharpness-Aware Minimization, SAM) 的研究成果。SAM是一种重要的神经网络正则化方法,通过在神经网络优化中添加对锐度概念的惩罚项来实现更好的泛化性能。最新的结果通过对锐度概念的明确,在理论层面揭示了SAM的内在机制。特别地,SAM中的两个近似步骤分别都会导致不准确的中间结果,但在全梯度方法下,它们的组合意外地表现了良好的效果。  来自德克萨斯大学奥斯汀分校的刘强助理教授展示了他在最优传输(Optimal transport)问题上取得的新结果。最优传输问题是在给定成本函数下最小化从一组数据传输到另一组数据的总成本的问题。通过一个简明的框架——整流(rectified flow),最优传输问题可以得到有效的求解。该算法只需要求解一系列非线性最小二乘优化问题,具有较高的计算效率。在实践中,通过该方法得到的ODE模型可以生成高质量的图像,且相较于扩散生成模型有显著的加速。此外,经过适当的修改,这一方法可以推广到高维连续分布的最优传输问题。
来自德克萨斯大学奥斯汀分校的刘强助理教授展示了他在最优传输(Optimal transport)问题上取得的新结果。最优传输问题是在给定成本函数下最小化从一组数据传输到另一组数据的总成本的问题。通过一个简明的框架——整流(rectified flow),最优传输问题可以得到有效的求解。该算法只需要求解一系列非线性最小二乘优化问题,具有较高的计算效率。在实践中,通过该方法得到的ODE模型可以生成高质量的图像,且相较于扩散生成模型有显著的加速。此外,经过适当的修改,这一方法可以推广到高维连续分布的最优传输问题。  斯坦福大学的Surya Ganguli教授在报告中展示了一种基于数据修剪的自监督算法。相比于以前的算法,基于数据修剪的方法更为简洁与节约,具有更强的可拓展性,且与当前最好的监督数据修剪算法具有相当的性能。进一步地,这一工作包含了网络规模的数据修剪,实验将已经高度筛选的440M LAION图像-文本对子集修剪到270M,而不会影响下游任务的准确性。因此,这一方法体现了合理的数据选择对机器学习模型性能提升的重要性。
斯坦福大学的Surya Ganguli教授在报告中展示了一种基于数据修剪的自监督算法。相比于以前的算法,基于数据修剪的方法更为简洁与节约,具有更强的可拓展性,且与当前最好的监督数据修剪算法具有相当的性能。进一步地,这一工作包含了网络规模的数据修剪,实验将已经高度筛选的440M LAION图像-文本对子集修剪到270M,而不会影响下游任务的准确性。因此,这一方法体现了合理的数据选择对机器学习模型性能提升的重要性。 微软亚洲研究院的董力研究员展示了他对大型预训练语言模型语境学习(In-Context Learning, ICL)能力的研究成果,提供了对ICL的性能优越性的一个理论解释。具体而言,可以将语言模型解释为元优化器,并将ICL理解为一种隐式调整。从理论上讲,可以证明Transformer的注意力机制具有梯度下降的隐式形式:GPT首先生成元梯度,然后应用其以构建ICL模型。实验结果也表明ICL的各项指标与显式的调整类似。此外,受此启发,一种基于动量梯度下降算法的注意力机制也得以提出。
微软亚洲研究院的董力研究员展示了他对大型预训练语言模型语境学习(In-Context Learning, ICL)能力的研究成果,提供了对ICL的性能优越性的一个理论解释。具体而言,可以将语言模型解释为元优化器,并将ICL理解为一种隐式调整。从理论上讲,可以证明Transformer的注意力机制具有梯度下降的隐式形式:GPT首先生成元梯度,然后应用其以构建ICL模型。实验结果也表明ICL的各项指标与显式的调整类似。此外,受此启发,一种基于动量梯度下降算法的注意力机制也得以提出。  微软研究院的Sebastien Bubeck研究员通过简单的稀疏编码模型,分析了Transformer中出现的Emergence现象。实验说明,训练该模型的神经网络时,在较大的学习率下,其收敛性质不稳定,会出现剧烈的振荡现象。分析表明,这一现象主要是由模型的非凸结构所导致。
微软研究院的Sebastien Bubeck研究员通过简单的稀疏编码模型,分析了Transformer中出现的Emergence现象。实验说明,训练该模型的神经网络时,在较大的学习率下,其收敛性质不稳定,会出现剧烈的振荡现象。分析表明,这一现象主要是由模型的非凸结构所导致。  上海交通大学的许志钦助理教授在报告中展示了他对于深度学习参数凝聚现象的研究。许老师首先提出了神经网络中的一个问题:为什么复杂的神经网络能够有好的拟合效果?在训练过程中,神经网络有两个隐性的规律,一个是频率原则,另一个是参数凝聚现象,后者作为非线性训练过程的特征,会使得训练样本的数量有效地降低。基于凝聚现象,许老师通过秩分析方法定量地理解了对于一个过参数化的神经网络,需要多大的数据集来保证其拟合效果较好。
上海交通大学的许志钦助理教授在报告中展示了他对于深度学习参数凝聚现象的研究。许老师首先提出了神经网络中的一个问题:为什么复杂的神经网络能够有好的拟合效果?在训练过程中,神经网络有两个隐性的规律,一个是频率原则,另一个是参数凝聚现象,后者作为非线性训练过程的特征,会使得训练样本的数量有效地降低。基于凝聚现象,许老师通过秩分析方法定量地理解了对于一个过参数化的神经网络,需要多大的数据集来保证其拟合效果较好。  东京大学的Masashi Sugiyama教授介绍了关于重要性加权方法(importance sampling method)的最新进展,并且将其用于解决分布偏移问题。在这次演讲中,他概述了在基于重要性加权的分布偏移问题的自适应方法,以及他最新的进展,包括对于协变量偏移的自适应估计,动态重要性加权对于联合分布偏移的自适应,以及多步骤类的先验偏移的自适应。
东京大学的Masashi Sugiyama教授介绍了关于重要性加权方法(importance sampling method)的最新进展,并且将其用于解决分布偏移问题。在这次演讲中,他概述了在基于重要性加权的分布偏移问题的自适应方法,以及他最新的进展,包括对于协变量偏移的自适应估计,动态重要性加权对于联合分布偏移的自适应,以及多步骤类的先验偏移的自适应。  北京大学的贺笛助理教授在报告中讨论了哪些图神经网络(GNN)能够用于解决实际问题。他的工作从一个本质不同的角度来研究GNN在Weisfiler-Lehman(WL)检验之外的功效。他引入了一种有效且具有原理的称为广义Weisfiler-Lehman距离检验(GD-WL),证明了这一检验对于具有双连通的度量都可行。实验表明他们的方法可以一致的优于先前的图神经网络结构。
北京大学的贺笛助理教授在报告中讨论了哪些图神经网络(GNN)能够用于解决实际问题。他的工作从一个本质不同的角度来研究GNN在Weisfiler-Lehman(WL)检验之外的功效。他引入了一种有效且具有原理的称为广义Weisfiler-Lehman距离检验(GD-WL),证明了这一检验对于具有双连通的度量都可行。实验表明他们的方法可以一致的优于先前的图神经网络结构。  清华大学的袁洋助理教授在报告中介绍了他关于对比学习的最新理论结果。对比学习是强大的自监督学习方法,但对于其理论性质的了解和对算法的理解相对缺乏。袁老师在文章中证明了对比学习和在相似图上谱聚类的等价性,并且强调这一等价性无需额外的条件。受这一理论的启发,在一些数据集上他们找到了比标准核函数效果更好的新的核函数。
清华大学的袁洋助理教授在报告中介绍了他关于对比学习的最新理论结果。对比学习是强大的自监督学习方法,但对于其理论性质的了解和对算法的理解相对缺乏。袁老师在文章中证明了对比学习和在相似图上谱聚类的等价性,并且强调这一等价性无需额外的条件。受这一理论的启发,在一些数据集上他们找到了比标准核函数效果更好的新的核函数。  新加坡国立大学的Kenji Kawaguchi教授在报告中介绍了对于Mixup方法的理论理解。Mixup 是一种流行的数据增强技术,用于训练深度神经网络,其通过线性插值输入对及其标签生成额外的样本,这种技术可以提高许多学习范式和应用程序中的泛化性能。
新加坡国立大学的Kenji Kawaguchi教授在报告中介绍了对于Mixup方法的理论理解。Mixup 是一种流行的数据增强技术,用于训练深度神经网络,其通过线性插值输入对及其标签生成额外的样本,这种技术可以提高许多学习范式和应用程序中的泛化性能。  香港大学的曹原助理教授在报告中介绍对两层卷积神经网络(CNN)的良性过拟合的研究。现代的神经网络模型经常可以在训练集过拟合的情况下仍然有较好的表现,这种现象被称为良性过拟合,但对于这种现象的理论层面的理解仍然比较缺乏。曹老师报告了他对于两层卷积神经网络的良性过拟合的研究,找到了一个信噪比需要满足的条件,使得CNN在该条件下为良性过拟合,反之则为有害的过拟合。
香港大学的曹原助理教授在报告中介绍对两层卷积神经网络(CNN)的良性过拟合的研究。现代的神经网络模型经常可以在训练集过拟合的情况下仍然有较好的表现,这种现象被称为良性过拟合,但对于这种现象的理论层面的理解仍然比较缺乏。曹老师报告了他对于两层卷积神经网络的良性过拟合的研究,找到了一个信噪比需要满足的条件,使得CNN在该条件下为良性过拟合,反之则为有害的过拟合。  来自北京大学的方聪助理教授在报告中介绍了关于环境不变的最小二乘问题。报告中考虑了一个多环境下的线性回归模型,该模型下的数据在不同的实验设定下采集。尽管该模型响应变量的联合分布、协方差可能在不同环境下不同,但其条件期望不变。他的工作构建了一个全新的,关于多环境不变的线性最小二乘(EILLS)目标函数,这一个最小二乘的多环境版本,利用上述条件期望不变结构以及不同环境之间的异质性来确定实际的参数。
来自北京大学的方聪助理教授在报告中介绍了关于环境不变的最小二乘问题。报告中考虑了一个多环境下的线性回归模型,该模型下的数据在不同的实验设定下采集。尽管该模型响应变量的联合分布、协方差可能在不同环境下不同,但其条件期望不变。他的工作构建了一个全新的,关于多环境不变的线性最小二乘(EILLS)目标函数,这一个最小二乘的多环境版本,利用上述条件期望不变结构以及不同环境之间的异质性来确定实际的参数。
最后,参会学者针对机器学习的理论基础展开进一步交流与讨论,会议通过线上及线下平台同时举办,来自全球近四百位学者参与本次会议。






 2023年2月,清华大学统计学研究中心侯琳副教授课题组在Nature Communications上发表了题为Quantifying portable genetic effects and improving cross-ancestry genetic prediction with GWAS summary statistics的研究论文[1],课题组2022届郭瀚民博士为该文的共同第一作者,侯琳副教授和威斯康辛大学麦迪逊分校吕琼石助理教授为该论文的共同通讯作者,威斯康辛大学麦迪逊分校的缪嘉诚为共同第一作者,宋格非、赵梓杰为本文的共同作者。
2023年2月,清华大学统计学研究中心侯琳副教授课题组在Nature Communications上发表了题为Quantifying portable genetic effects and improving cross-ancestry genetic prediction with GWAS summary statistics的研究论文[1],课题组2022届郭瀚民博士为该文的共同第一作者,侯琳副教授和威斯康辛大学麦迪逊分校吕琼石助理教授为该论文的共同通讯作者,威斯康辛大学麦迪逊分校的缪嘉诚为共同第一作者,宋格非、赵梓杰为本文的共同作者。